
Medical devices support the examination, diagnosis, treatment, and follow-up knowledge of the doctor or surgeon. Having access to medical equipment enables a doctor to accurately diagnose a patient’s illness or a surgeon to perform a procedure.
These devices are just as important as medications, and they go through the same clinical investigation process as well, including pilot, pivotal, and post-approval studies. Therefore, clinical investigations are carried out globally by a variety of medical device manufacturers before the introduction of their devices in the medical device industry (pharma), not only by pharmaceutical companies for the approval of their devices.
In the past, studies were needed if the studies of new medical devices conform to one of the 14 categories that had been alerted. This has changed as a result of the MDR, and on November 1, 2017, relating to the 2019 Medical Devices (Second Amendment) Rules. To safeguard human health by ensuring the efficacy and safety of each new medical device prior to its release on the market, rules and regulations for clinical trials of new drugs and medical devices are periodically amended.
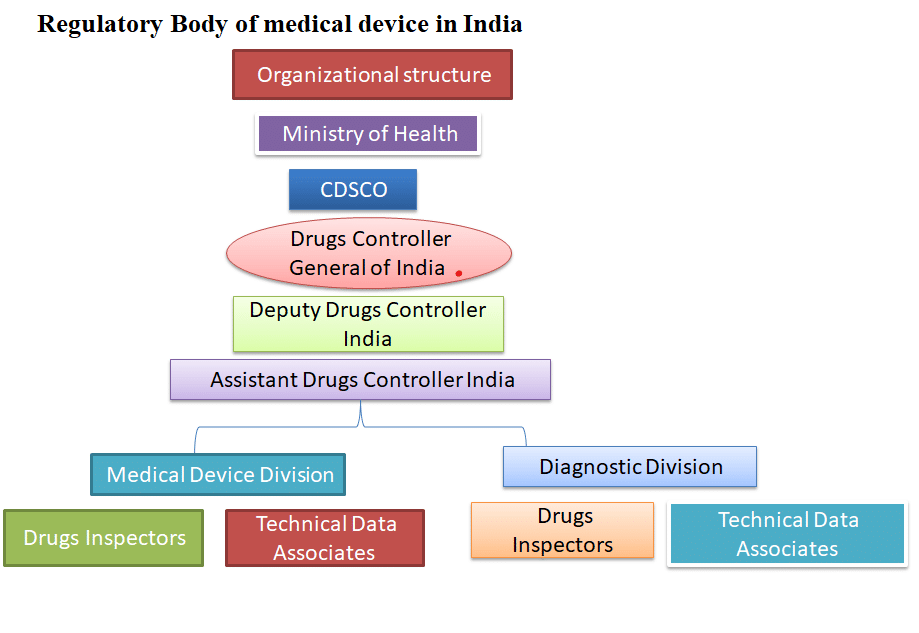
The list includes 351 medical devices divided into 29 categories , as well as 247 IVDs divided into 21 categories. a new list of notified medical devices was published. Without the approval of the CDSCO and the ethical committee, no sponsor may conduct a clinical investigation. The Sponsor/CRO submits an online application to conduct a pilot or pivotal clinical investigation in Form MD-22 and permission grant conduct a clinical investigation of medical devices in form MD-23 and MD-24 for IVD application clinical investigation and MD-25 are conducted in clinical investigation, along with the information specified in the MDR’s Seventh Schedule. The pivotal study requires data from pilot clinical investigation as well. The clinical investigation should begin within one year of the grant of permission by submitting the first participant; otherwise, specific approval from CDSCO is allowed to start the clinical investigation.
Before the first patient is enrolled, every study must be registered with the Clinical Trial Registry of India. Furthermore, the sponsor must submit to CDSCO annually the status of the study data (open, completed, or terminated).
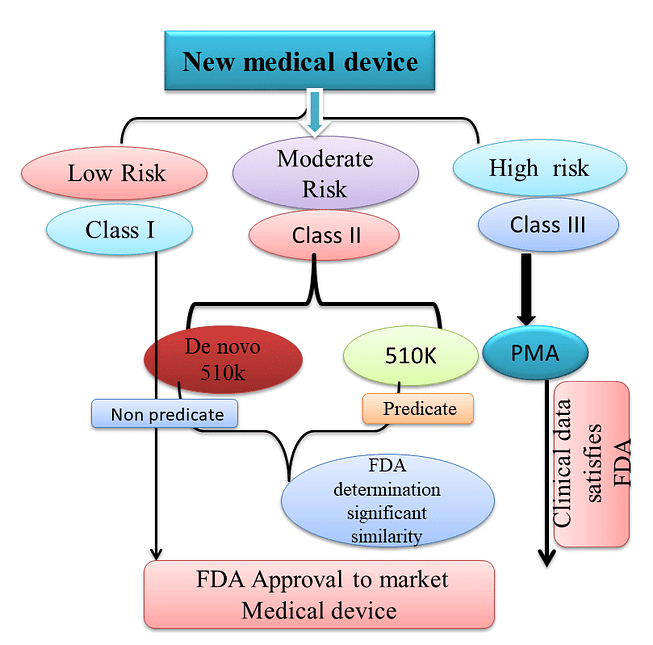
The FDA regulates the implementation of the second set of regulations in the United States. Some relatively simple devices are exempt from the FDA’s clearance requirement and can be marketed in the United States without prior authorization. Commercial distribution of the residual medical devices usually requires FDA marketing approval.
The applicant of the Form MD-27 (to be filled before releasing the medical device in the market)must notify the Central Licensing Authority(CLA) the date of introduction of the medical device to the market and must submit periodic safety update reports (PSURs) starting from that date. The PSURs must first be submitted every six months for the first two years, after which they must be submitted annually for the next two years.
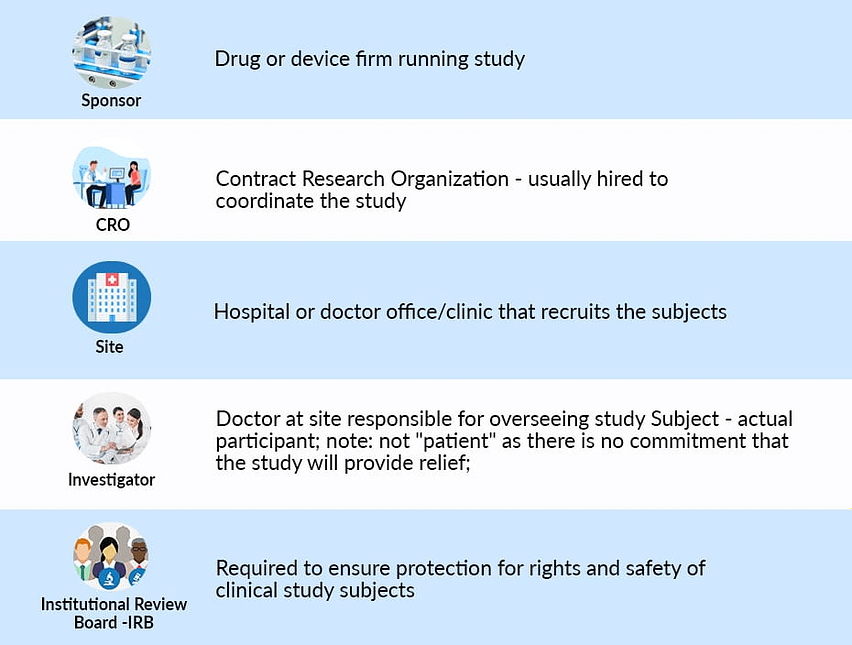
A clinical investigation (medical device) is successful if it produces scientifically reliable clinical data, comes to medically and scientifically sound study conclusions, and is finished on schedule. This means that all parties involved in the clinical investigation like sponsors, investigators, clinical data monitors, and others, must fully understand and comply with all regulatory requirements, standards, and documents.
Reference
A Risk-Based Approach to Monitoring of Clinical Investigations Questions and Answers Guidance for Industry (Food and Drug Administration Draft Guidance, March 2019).